

Subscript
1. Introduction
2. Results and Discussion
2.1 NFA Design and Material Properties
2.2 Photovoltaics Performance
3. Experimental Details
3.1 Materials and Measurements
3.2 Synthetic Procedure Details
4. Conclusions
Subscript
OSCs : Organic solar cells
NFAs : non-fullerene acceptors
CPDT : cyclopentadithiophene
BT : benzothiadiazole
NIR : near-infrared
IDT : indaceno[1,2-b:5,6-b′]dithiophene
PCE : power conversion efficiency
UV–Vis : Ultraviolet–visible
1H NMR : 1H nuclear magnetic resonance spectroscopy
VOC : open-circuit voltage
FF : fill factor
o-XY : ortho-xylene
MALDI-TOF : matrix-assisted laser desorption/ionization time-of-flight
ICT : intramolecular charge transfer
EQE : external quantum efficiency
µe : electron mobility
Jph : photo-current density
JSC : short-circuit current density
DSC : differential scanning calorimetry
TGA : thermogravimetric analysis
HOMO : highest occupied molecular orbitals
LUMO : lowest unoccupied molecular orbitals
CV : Cyclic voltammetry
Td : decomposition temperature
Tm : melting temperature
1. Introduction
Organic solar cells (OSCs) have garnered significant attention recently due to their potential for low-cost, flexible, and environmentally friendly energy solutions. The active layer materials, particularly non-fullerene acceptors (NFAs), are central to the performance of OSCs, which have evolved as promising alternatives to traditional fullerene-based acceptors1-5). A key focus of ongoing research is developing novel NFAs that can deliver enhanced light absorption, higher charge mobility, and better compatibility with various donor polymers. Compared to traditional fullerene-based systems, OSCs utilizing NFAs can simultaneously achieve higher open-circuit voltage (Voc) and short-circuit current density (Jsc). Additionally, structural modifications of the side chains, terminal groups, and rigid ladder-type cores have been shown to effectively fine-tune the bandgaps, energy levels, and packing properties of NFAs6-9). In this regard, Y6, a non-fullerene acceptor, has gained prominence for its impressive OSC performance. Among various NFAs, Y6-based materials have emerged as a groundbreaking class due to their unique molecular structure and superior photovoltaic properties10-13).
Over time, an increasing number of Y6 derivatives have been synthesized, utilizing a range of modification strategies. These modifications can be broadly categorized into two main approaches: symmetric and asymmetric. The advantages of asymmetric modifications include providing adjustable optoelectronic characteristics (e.g., molecular dipole moment and intermolecular binding energy) while partially preserving the original molecular structure14). The asymmetric modification of Y6 has mainly been categorized into three aspects: end groups, core moieties, and side-chains15). Among those, the conjugated central core is a key component of NFAs, as it determines their primary physical properties, including absorption characteristics, energy levels, and inter- and intramolecular interactions. Specifically, an asymmetric extension of central core units can change the above-mentioned properties while maintaining relative π-electron delocalization16, 17). Asymmetric alteration of the length/shape of the conjugated backbone significantly influences the molecular conformations. Also, increasing the number of fused rings readily shifts the absorption towards the near-infrared (NIR) wavelength region, which significantly enhances photocurrent generation and ultimately impacts the JSC and PCE of OSCs7, 17-24). For instance, the typical indaceno[1,2-b:5,6-b′]dithiophene (IDT) unit can be transformed into an asymmetric backbone by incorporating additional conjugated fused ring on one side. The Sun group utilized this approach to design three distinct fused-ring acceptors: TPT-2F, TPTT-2Fm, and TPTTT-2F. This extended IDT backbone led to broader absorption spectra and elevated energy levels. The wider absorption and elevated energy levels were achieved by the extended IDT backbone17). In addition, the Zou group developed three types of fused ring acceptors, Y26, Y22, and Y18, featuring five-, six-, and seven-membered conjugated fused rings, respectively25-28). The six-membered asymmetric acceptor Y22 demonstrated significantly higher efficiency than that of the five-membered symmetric acceptor Y26. This improvement can be attributed to the asymmetric expansion of molecular structure. Extending the thiophene conjugation progressively resulted in red-shifted absorption spectra and enhanced intermolecular ordering, leading to stronger π–π interactions. These findings provide insight into the potential development of asymmetric Y6-based NFA.
Herein, enlightened by the attractive features of aforementioned asymmetric extension strategies, the π-conjugation of Y6 was further extended by replacing thieno[3,2-b]thiophene (TT), through the fusion of cyclopentadithiophene (CPDT) unit on one side of Y6 core, to construct a novel fused octacyclic acceptor OCPDTBT. Note that CPDT has been widely used as a building block to construct conjugated polymers29-32), and have recently been employed to construct NFAs33, 34), as they usually present relatively small bandgaps and red-shifted the absorptions35-39). Therefore, Iin this study, we present a series of newly designed asymmetric octacylic NFAs, incorporating a fused CPDT, benzothiadiazole (BT), and TT as the central core, and IC-2F as the electron-withdrawing end groups. To further fine-tune the material characteristics, side-chain modifications were performed, aiming at structural optimization of the NFAs. We observed that compared to the well-established Y6 derivatives, the newly synthesized OCPDTBT NFAs exhibit a red-shifted and broader absorption spectra extending to a wavelength of ~988 nm. These improved optical and electronic properties are attributed to the extended conjugation and strong electron-donating capability introduced by the CPDT core. As a result, the PM6:OCPDTBT-HD blend-based OSCs processed with a non-halogenated solvent (i.e., o-xylene) demonstrated a high PCE of 13.47%, with a high JSC of 24.17 mA cm-2 and an impressive FF of 71%. These results highlight the potential of OCPDTBT NFAs as a promising class of materials for low-bandgap OSCs, particularly in environmentally benign processing conditions. This study also offers new insights into the potential of OCPDTBT acceptors for next-generation organic photovoltaics.
2. Results and Discussion
2.1 NFA Design and Material Properties
The chemical structures and synthetic route of OCPDTBT-based NFAs are shown in Fig. 1. The NFAs were designed by incorporating a fused CPDT, BT, and TT moieties as the central core. Side-chain modifications were performed by introducing 2-hexyldecyl (HD), 2-octyldodecyl (OD), and 2-decyltetradecyl (DT) at pyrrole position. Their successful synthesis was confirmed by 1H nuclear magnetic resonance spectroscopy (1H NMR) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) measurements.
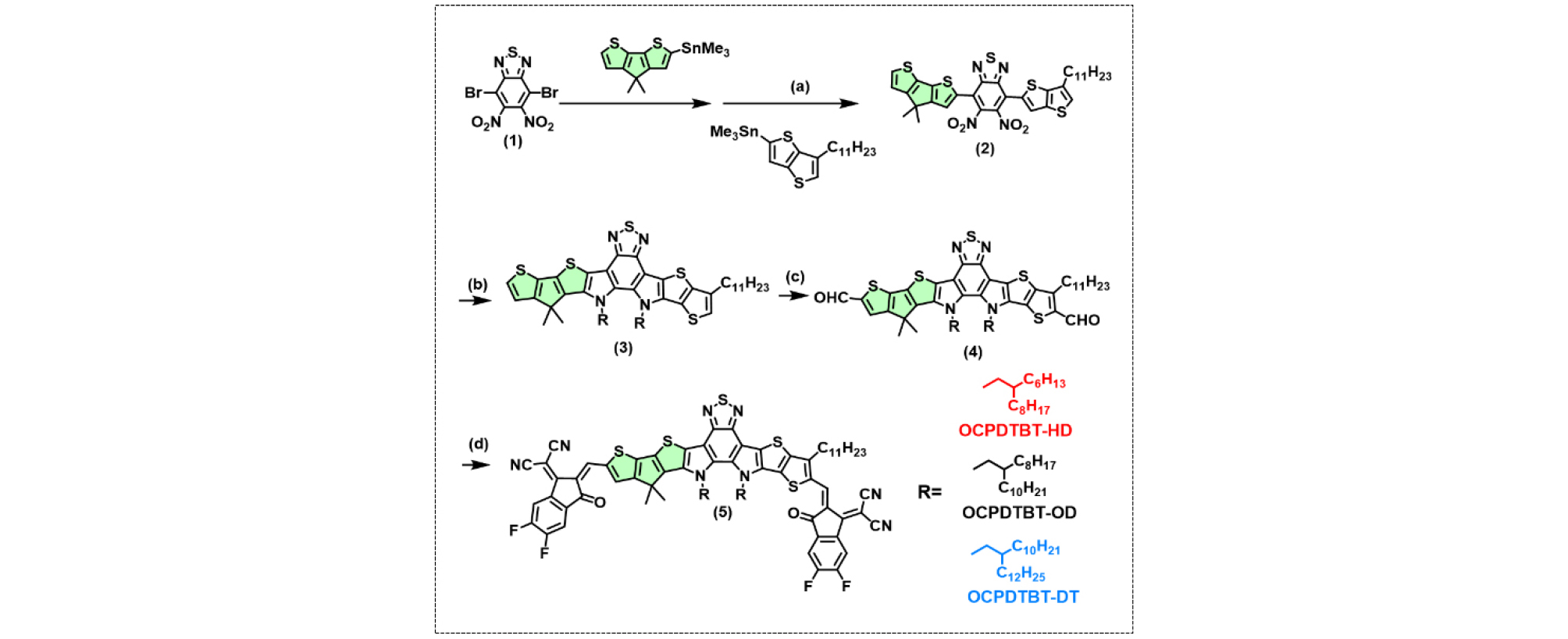
Ultraviolet–visible (UV–Vis) absorption spectroscopy in chloroform solution and neat films was performed to investigate the optical properties of OCPDTBT-HD, OCPDTBT-OD, and OCPDTBT-DT (Fig. 2a, b and Table 1). In solution, all the NFAs showed similar wide and strong absorption in the 640–810 nm wavelength region and nearly identical maximum absorption peak. In films, the absorption spectra of the NFAs were significantly red-shifted compared to their solution spectra, indicating substantial aggregation in the solid state. In addition, by increasing the alkyl chain length from HD to DT, a blue-shifted absorption was observed in the film, as confirmed by the change in the absorption onset (λonset) from 988 to 958 nm. This result might be due to the increasing steric hindrance with the increase in alkyl chain size, which can weaken the π–π stacking interactions between NFA molecules in solid state. Reduced π–π stacking leads to decreased molecular overlap, weakening the electronic interactions between adjacent molecules. This results in less delocalization of π-electrons, causing the absorption spectrum to shift toward higher energy (blue shift)40-42). Furthermore, the newly designed NFAs exhibited red-shifted absorption compared to the Y6 derivatives, with OCPDTBT-HD achieving λmaxfilm = 853 nm and λonset = 988 nm. The enhanced absorption of these NFAs is due to the increased conjugation and strong electron donating property of CPDT-based core. The optical bandgaps (Egopts) of OCPDTBT-HD, OCPDTBT-OD, and OCPDTBT-DT were estimated to be 1.25, 1.27, and 1.29 eV respectively, from the λonset values of thin films. These results show that extending conjugation length of core significantly red-shifts the absorption spectrum and enhance the intramolecular charge transfer (ICT).
Table 1.
Optical and electrochemical properties of the OCPDTBT NFAs
| NFA |
λmaxsoln [nm]a |
λmaxfilm [nm]b |
λonset [nm] | Egopt [eV]c | EHOMO [eV]d | ELUMO [eV]d |
| OCPDTBT-HD | 770 | 853 | 988 | 1.25 | −5.58 | −4.09 |
| OCPDTBT-OD | 770 | 843 | 974 | 1.27 | −5.59 | −4.07 |
| OCPDTBT-DT | 770 | 850 | 958 | 1.29 | −5.60 | −4.05 |
Cyclic voltammetry (CV) was performed to estimate the energy levels of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of the NFAs (Fig. 2c and Table 1). The HOMO/LUMO energy levels of OCPDTBT-HD, OCPDTBT-OD, and OCPDTBT-DT were measured to be −5.58/−4.09, −5.59/−4.07, and −5.60/−4.05 eV, respectively, showing well-matched energy level with the PM6 donor.
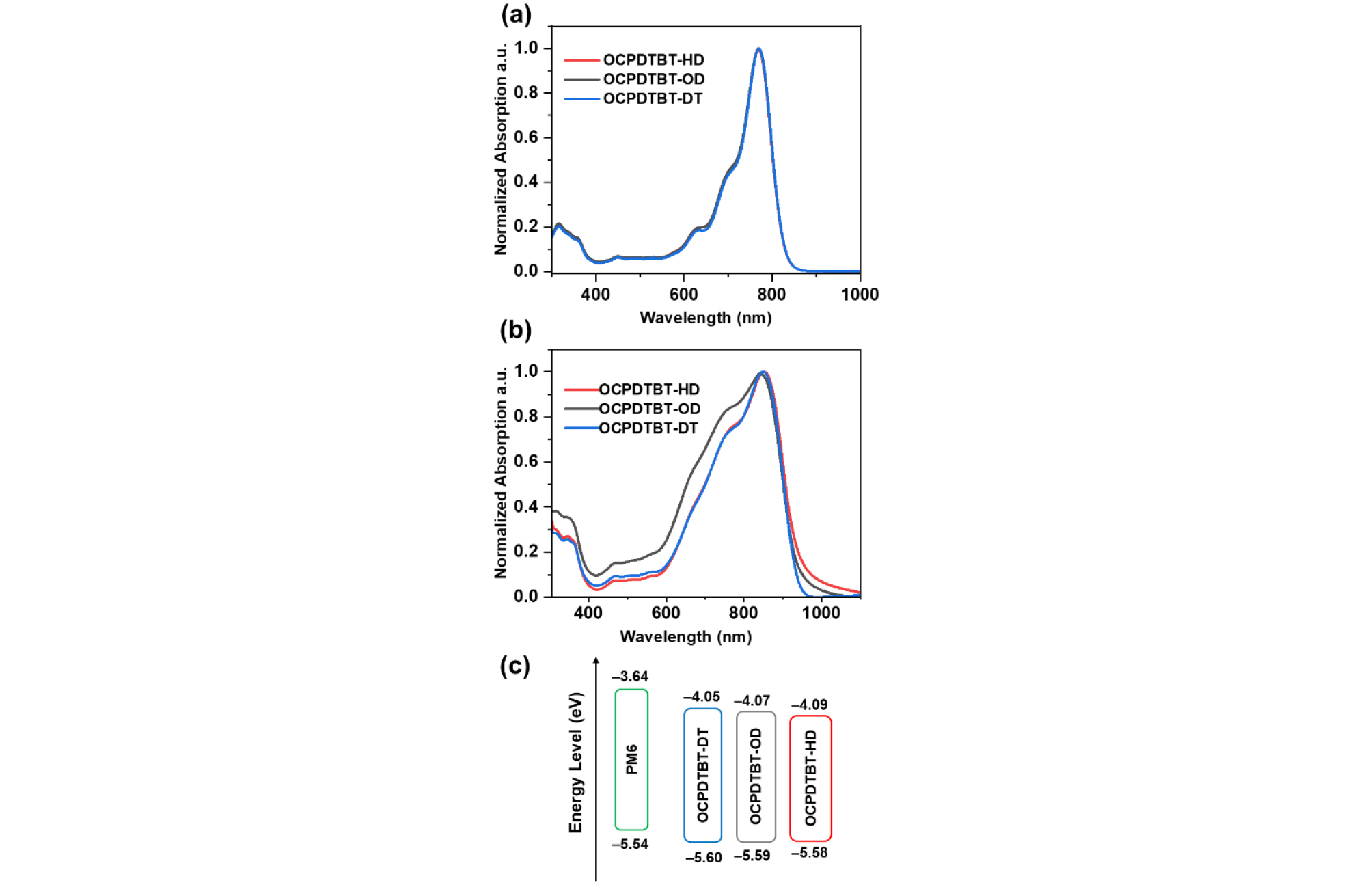
The thermal properties of the NFAs were characterized by thermogravimetric analysis (TGA) (Fig. 3a). The thermal decomposition temperatures (Td) with 5% weight loss were 343.9°C, 333.4°C, and 32.0°C, for OCPDTBT-HD, OCPDTBT-OD and OCPDTBT-DT, respectively. These results indicate that all the compounds have good thermal stability for OSCs application. Differential scanning calorimetry (DSC) measurements were conducted to investigate the crystalline properties of the NFAs (Fig. 3b). All the compounds exhibited clear melting behaviors. As the size of the alkyl chain increased, the melting temperature (Tm) of NFAs was gradually reduced. The Tm was 235.3°C, 229.3°C, and 217.3°C for OCPDTBT-HD, OCPDTBT-OD and OCPDTBT-DT, respectively. The decrease in Tm according to the size of the alkyl chain can be interpreted as weakened intermolecular interactions resulting from the steric hindrance effect of longer alkyl chains40, 43).
2.2 Photovoltaics Performance
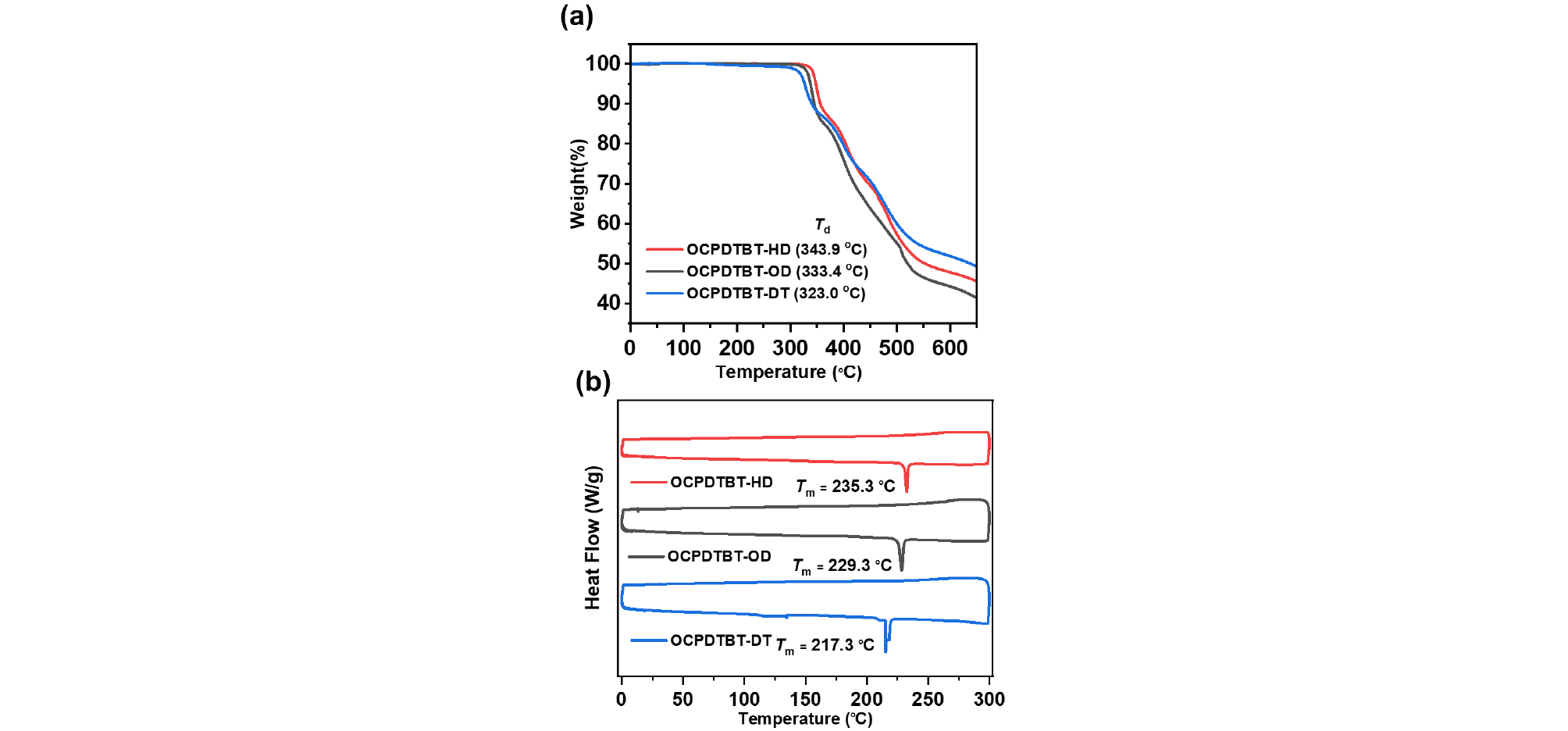
To analyze the influence of asymmetric OCPDTBT NFAs on device performance, OSCs were fabricated using a non-halogenated ortho-xylene (o-XY) solvent. The device structure used in the device fabrication was indium tin oxide (ITO)/ ZnO nanoparticles (ZnO NPs)/ poly(ethylenimine) ethoxylated (PEIE)/ photoactive layer/ MoO3/ Ag. The current density–voltage (J–V) characteristics of the OSC devices are illustrated in Fig. 4a, and the corresponding photovoltaic metrics are listed in Table 2.
Table 2.
Summary of device parameters of the optimized OSCs based on PM6:NFAs
| NFA | VOC (V) | JSC (mA cm-2) |
FF (%) |
PCE (%) |
| PM6:OCPDTBT-HD | 0.79 | 24.17 | 70.97 | 13.47 |
| PM6:OCPDTBT-OD | 0.81 | 21.31 | 71.00 | 12.30 |
| PM6:OCPDTBT-DT | 0.83 | 17.42 | 64.47 | 9.41 |
For PM6:OCPDTBT-HD OSCs with HD side chain, the device delivered the highest performance (PCE = 13.47%, VOC = 0.79 V, JSC = 24.17 mA cm-2, and FF = 71%) among the series. However, further increase in the side-chain length linearly decreased the photovoltaic performance. For instance, the PCEs of PM6:OCPDTBT-OD and PM6:OCPDTBT-DT were 12.3% and 9.41%, respectively. The reduced performance is mainly attributed to the drop in both JSC (24.17 → 17.42 mA cm-2) and FF (71 → 64%). The lower JSC of PM6:OCPDTBT-DT-based OSCs is associated with reduced π–π stacking and blue-shifted EQE spectra. By contrast, the superior performance of the PM6:OCPDTBT-HD-based device is likely attributed to its optimized active layer morphology and better light harvesting ability. The EQE spectrum of OSC based on PM6:OCPDTBT-HD showed a peak value approaching 80% (Fig. 4b). These results suggest that slight modification of the alkyl chain length of NFAs has a major impact on the overall performance of OSCs processed by a non-halogenated solvent. In addition, all the OSCs exhibited broad photon responses from 300 to 980 nm, consistent with the UV–vis absorption spectra of the blend films.
3. Experimental Details
3.1 Materials and Measurements
All commercially available reagents and solvents were directly used without further purification unless otherwise specified. 1H NMR (400 MHz) spectra were recorded on Bruker AVANCE III HD 400 MHz NMR spectrometer in CDCl3. The MALDI-TOF mass spectra were obtained by Bruker Autoflex instrument. TGA was performed using Thermo plus EVOII TG8120, samples were run under N2 and heated from room temperature to 600°C at a rate of 10°C min-1. UV–Vis absorption measurements were carried out with a double-beam Shimadzu UV-2550 spectrophotometer over 300–1000 nm. The UV–Vis spectra in solution were obtained in dilute chloroform solution, while the UV–Vis spectra in thin film were obtained by spin-coating chloroform solution (15 mg mL-1) onto quartz substrates. CV was measured by using an IviumStat instrument and conducted at a scan rate of 50 mV s-1 at 25°C under argon with 0.1 M tetrabutylammonium hexafluorophosphate in acetonitrile as the electrolyte. The NFAs were deposited onto the working electrode from chloroform solution. All CV measurements were carried out at room temperature with a conventional three-electrode configuration employing a glassy carbon electrode as the working electrode, a saturated calomel electrode as the reference electrode, and a Pt wire as the counter electrode. The HOMO and LUMO energy levels were calculated from the onset oxidation and reduction potential, respectively, using the empirical relationship EHOMO = −(Eonset,ox − E1/2,ferrocene + 4.8) eV and ELUMO = −(Eonset,red − E1/2,ferrocene + 4.8) eV. The OSC structure was ITO/ ZnO NPs/ PEIE/ photoactive layer/ MoO3/ Ag. The pre-patterned ITO/glass substrates were ultrasonicated in the order of detergent, acetone, and isopropanol. After being dried at around 140°C in an oven for 4 h, the substrates were then treated by UV-ozone exposure for 15 min. Subsequently, a thin layer of ZnO NPs (30 nm) in butanol was deposited at 3000 rpm for 30 s and baked at 110 °C for 10min. Then, PEIE solution was prepared by dissolving PEIE in methoxyethanol at weight concentration of 0.1 wt%. A layer of PEIE (5 nm) was then deposited onto the ZnO NPs at 5000 rpm for 40 s and then annealed at 110 °C for 10 min. The ITO/ZnO NPs/PEIE substrates were transferred to an N2-filled glovebox for deposition of the photoactive layer. The PM6:assymetric derivatives (1.0:1.2 w/w) solutions were spin cast on top of the PEIE layer at 2500 rpm from an o-xylene solvent only. Afterwards, the films were annealed at 130°C for 10 min. Finally, MoO3 (10 nm) and Ag (100 nm) were deposited on top of the photoactive layer in a high vacuum chamber (~10-6 torr). The photovoltaic properties of OSCs were evaluated through a photomask with an aperture size of 0.09 cm2.
The PCE and EQE of OSCs were characterized using Keithley 2400 source meter and a solar simulator (K201 LAB55, McScience), under simulated 100 mW cm-2 irradiation from a Xe arc lamp with an AM 1.5G filter. Simulator irradiance was characterized using a calibrated spectrometer and the illumination intensity was set using an NREL-certified silicon diode with an integrated KGI optical filter. EQEs were measured using a spectral measurement system (K3100 IQX, McScience Inc.) which applied monochromatic light from the 100 W Xe arc lamp filtered by an optical chopper and a monochromator. All the blend films were processed using o-xylene without any additive.
3.2 Synthetic Procedure Details
The synthetic routes towards assymetric non-fullerene acceptors are shown in Fig. 1 Compound trimethyl(6-undecylthieno[3,2-b]thiophen-2-yl)stannane, trimethyl(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)stannane were synthesized by the procedure in literature44) followed by the coupling reaction with compound 1 to yield compound 2. Cyclization of compound 2 using triethylphosphite, the reaction with R-Br under alkaline conditions. The Vilsmeier-Haack reaction of compound 3 with POCl3 and DMF generated compound 4, which was further converted to the desired compound 5 via the Knoevenagel condensation reaction of compound 4 with 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1 ylidene) malononitrile.
Synthesis of compound 2: To a dry 100 ml round-bottom flask, compound 1 (2 g, 5.20 mmol), trimethyl(6-undecylthieno[3,2-b]thiophen-2-yl)stannane (3.09 g, 6.77 mmol), trimethyl(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)stannane (2.49 g, 6.77 mmol) Pd(PPh3)2Cl2 (182 mg, 0.26 mmol) and dry THF (30 mL) was added under argon protection. Then, the mixture was refluxed overnight at 90°C. After cooling to room temperature, the solvent was evaporated and the residue was purified with column chromatography on silica gel using a mixture of solvents hexane and dichloromethane 5:1 as an eluent to give compound 2 as a purple sticky solid (1.45 g, 38%). 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.48 (s, 1H), 7.33-7.35 (d, 1H), 7.15 (s, 1H), 7.04-7.05 (d, 1H), 2.75-2.79 (t, 2H), 1.75-1.82 (m, 2H), 1.43-1.27 (m, 16H), 0.86-0.90 (m, 9H).
Synthesis of compound 3: To a dry 25 ml round-bottom flask, compound 2 (0.5 g, 0.69 mmol), triethyl phosphite (2.5 mL) and anhydrous o-dichlorobenzene (10 mL) was added under argon protection. Then, the mixture was refluxed overnight at 180°C. After cooling to room temperature, the solvent was evaporated under reduced pressure. Without further purification, R-Br (4.15 mmol), potassium iodide (46 mg, 0.27 mmol), potassium carbonate (0.95 g, 6.9 mmol) and N, N-dimethylformamide (30 mL) were added into the residue. The mixture was kept stirring overnight at 90°C. After cooling to room temperature, the mixture was extracted with dichloromethane and washed with water. The collected organic layer was dried over anhydrous MgSO4, filtrated, and concentrated under reduced pressure. The residue was purified with column chromatography on silica gel using a mixture of solvent hexane and dichloromethane 4:1 as an eluent to give compound 3.
Compound HD: The starting material compound R-Br should be compound 2-hexyl decyl bromide (1.27 g, 4.15 mmol). The final product compound was obtained as a yellow sticky solid (280 mg, 37%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7.20-7.21(d, 1H), 7.05-7.06 (d, 1H), 7.00 (s, 1H), 4.52-4.69 (m, 4H), 2.80-2.84 (t, 2H), 1.84-2.01 (m, 4H), 1.72-1.84 (m, 6H), 1.26- 1.45 (m, 39H), 0.90-1.22 (m, 28H), 0.78-0.89 (m, 12H).
Compound OD: The starting material compound R-Br should be compound 2-octyl dodecyl bromide (1.49 g, 4.15 mmol). The final product compound was obtained as a yellow sticky solid (300 mg, 36%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7.20-7.21(d, 1H), 7.04-7.05 (d, 1H), 7.00 (s, 1H), 4.52-4.69 (m, 4H), 2.80-2.84 (t, 2H), 1.84-2.01 (m, 4H), 1.72-1.84 (m, 6H), 1.26-1.45 (m, 49H), 0.90-1.22 (m, 34H), 0.77-0.89 (m, 12H).
Compound DT: The starting material compound R-Br should be compound 2-decyltetradecyl bromide (1.73 g, 4.15 mmol). The final product compound was obtained as a yellow sticky solid (330 mg, 35%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7.19-7.20 (d, 1H), 7.05-7.06 (d, 1H), 7.00 (s, 1H), 4.52-4.69 (m, 4H), 2.80-2.84 (t, 2H), 1.84-2.01 (m, 4H), 1.72-1.84 (m, 6H), 1.26-1.45 (m, 59H), 0.90-1.22 (m, 40H), 0.75-0.89 (m, 12H).
Synthesis of compound 4: To a solution of 3 (0.25 mmol) in 1,2-dichloroethane (15 mL), DMF (0.4 mL) was added under the protection of argon. The reaction mixture was then cooled to 0°C, followed by slow addition of POCl3 (1 mL). After being kept at 0°C for 1 h, the reaction mixture was heated to 70°C and stirred for 6 h. After cooled to room temperature, the reaction mixture was quenched with NaHCO3 solution and extracted with dichloromethane. The combined extracts were washed with brine, dried over anhydrous MgSO4, and then filtered. The solvent was removed by rotary evaporation to yield the crude product. The obtained crude product was further purified by column chromatography on silica gel with a mixture solvent of hexane and dichloromethane 1:1 as an eluent to afford compound 4.
Compound HD: The starting material compound 3 should be compound HD (0.28 g, 0.25 mmol). The final product compound was obtained as a bright yellow solid (0.150 g, 51%). 1H NMR (400 MHz, CDCl3, δ): 10.14 (s, 1H), 9.86 (s, 1H), 7.69 (s, 1H), 4.53-4.72 (m, 4H), 3.18-3.22 (t, 2H), 1.92-2.03 (m, 4H), 1.79-1.75 (m, 6H), 1.26-1.46 (m, 39H), 0.90-1.22 (m, 28H), 0.79-0.90 (m, 12H)
Compound OD: The starting material compound 3 should be compound OD (0.30 g, 0.25 mmol). The final product compound was obtained as a bright yellow solid (0.164 g, 52%). 1H NMR (400 MHz, CDCl3, δ): 10.14 (s, 1H), 9.86 (s, 1H), 7.69 (s, 1H), 4.54-4.71 (m, 4H), 3.18-3.22 (t, 2H), 1.97-2.02 (m, 4H), 1.95-1.75 (m, 6H), 1.26-1.48 (m, 49H), 0.90-1.22 (m, 34H), 0.77-0.90 (m, 12H).
Compound DT: The starting material compound 3 should be compound DT (0.33 g, 0.25 mmol). The final product compound was obtained as a bright yellow solid (0.170 g, 54%). 1H NMR (400 MHz, CDCl3, δ): 10.14 (s, 1H), 9.86 (s, 1H), 7.68 (s, 1H), 4.52-4.72 (m, 4H), 3.18-3.21 (t, 2H), 1.92-1.95 (m, 4H), 1.74-1.80 (m, 6H), 1.26-1.49 (m, 59H), 0.90-1.22 (m, 40H), 0.86-0.90 (m, 12H).
Synthesis of compound 5: Under nitrogen protection, compound 4 (0.12 mmol), 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (110 mg, 0.48 mmol) and chloroform (20 mL) was added to 50 mL two-necked round bottom flask. After the reaction mixture was stirred at room temperature for 10 mins, pyridine (0.4 mL) was then added. The reaction mixture was allowed to stir at 65°C for 8 h. After removal of solvent of reaction mixture, methanol was added and the precipitate was collected by filtration to get crude product, which was further purified by column chromatography on silica gel with hexane: dichloromethane (1:1) as an eluent to afford compound 5.
Compound HD: The starting material compound 4 should be compound HD (140 mg, 0.12 mmol). The final product compound OCPDTBT-HD was obtained as a dark solid (100 mg, 52%). 1H NMR (400 MHz, CDCl3, δ): 9.17 (s, 1H), 8.92 (s, 1H), 8.51-8.60 (m, 2H), 7.64-7.75 (m, 3H), 4.50-4.73 (m, 4H), 3.21-3.25 (t, 2H), 1.87-2.01 (m, 4H), 1.81-1.84 (m, 6H), 1.26-1.42 (m, 39H), 0.95-1.25 (m, 28H), 0.78-0.90 (m, 12H). MS (MALDI-TOF) m/z: calcd 1586.67, found M+: 1586.68.
Compound OD: The starting material compound 4 should be compound OD (155 mg, 0.12 mmol). The final product compound OCPDTBT-OD was obtained as a dark solid (100.2 mg, 49%). 1H NMR (400 MHz, CDCl3, δ): 9.17 (s, 1H), 8.90 (s, 1H), 8.50-8.60 (m, 2H), 7.61-7.73 (m, 3H), 4.50-4.73 (m, 4H), 3.21-3.25 (t, 2H), 1.87-2.01 (m, 4H), 1.81-1.84 (m, 6H), 1.26-1.42 (m, 49H), 0.95-1.25 (m, 34H), 0.80-0.90 (m, 12H). MS (MALDI- TOF) m/z: calcd 1698.79, found M+: 1700.
Compound DT: The starting material compound 4 should be compound DT (168 mg, 0.12 mmol). The final product compound OCPDTBT-DT was obtained as a dark solid (100.5 mg, 46%). 1H NMR (400 MHz, CDCl3, δ): 9.17 (s, 1H), 8.91 (s, 1H), 8.51-8.61 (m, 2H), 7.63-7.74 (m, 3H), 4.49-4.73 (m, 4H), 3.21- 3.25 (t, 2H), 1.88-1.99 (m, 4H), 1.81-1.87 (m, 6H), 1.26-1.38 (m, 59H), 0.95-1.25 (m, 40H), 0.81-0.90 (m, 12H). MALDI-TOF-MS m/z: calcd. 1810.92, found M+: 1810.62
4. Conclusions
In conclusion, this study demonstrates the successful design and synthesis of a novel class of asymmetric A-DA'D-A type octacyclic NFAs based on a CPDT and BT core units, and IC-2F end groups. These molecular modifications result in red-shifted and broader absorption, extending the absorption onset to 988 nm. The incorporation of side-chain modifications effectively fine-tuned the electronic and structural properties. The remarkable photovoltaic performance, evidenced by a PCE of 13.47%, a high JSC of 24.17 mA cm-2, and a fill factor of 71%, was achieved with PM6:OCPDTBT-HD blends processed in non-halogenated solvent (o-XY). The findings reveal that OCPDTBT NFAs are a highly promising material class for low-bandgap OSCs, especially when processed under eco-friendly conditions. This work also provides valuable insights into the application of OCPDTBT acceptors in advancing next-generation organic photovoltaics.
